


Aurich Lawson
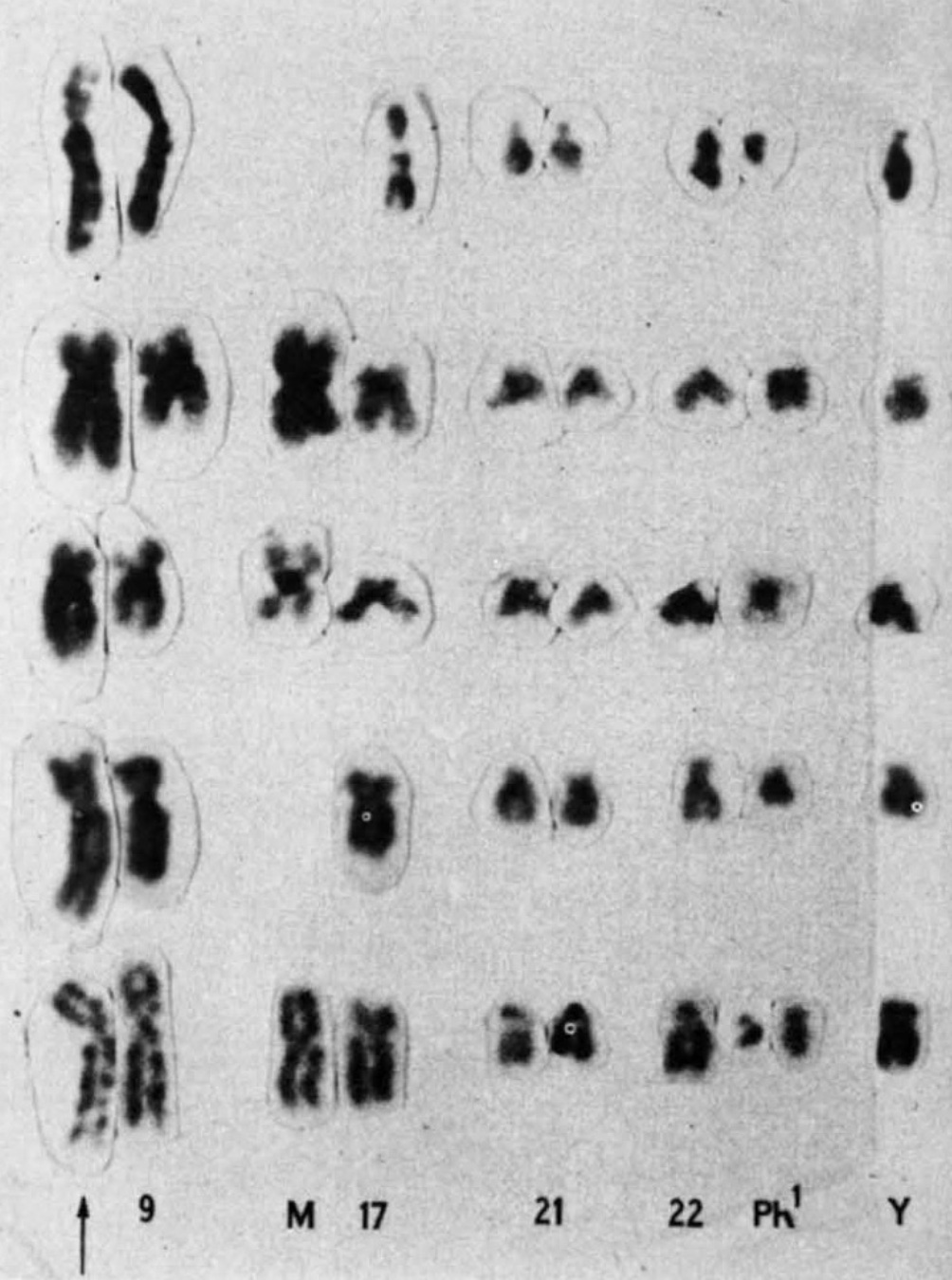
In 1972, Janet Rowley sat at her dining room table and cut tiny chromosomes from photographs she had taken in her laboratory. One by one, she snipped out the small figures her children teasingly called paper dolls. She then carefully laid them out in 23 matching pairs—and warned her kids not to sneeze.
The physician-scientist had just mastered a new chromosome-staining technique in a year-long sabbatical at Oxford. But it was in the dining room of her Chicago home where she made the discovery that would dramatically alter the course of cancer research.

{kind=link}
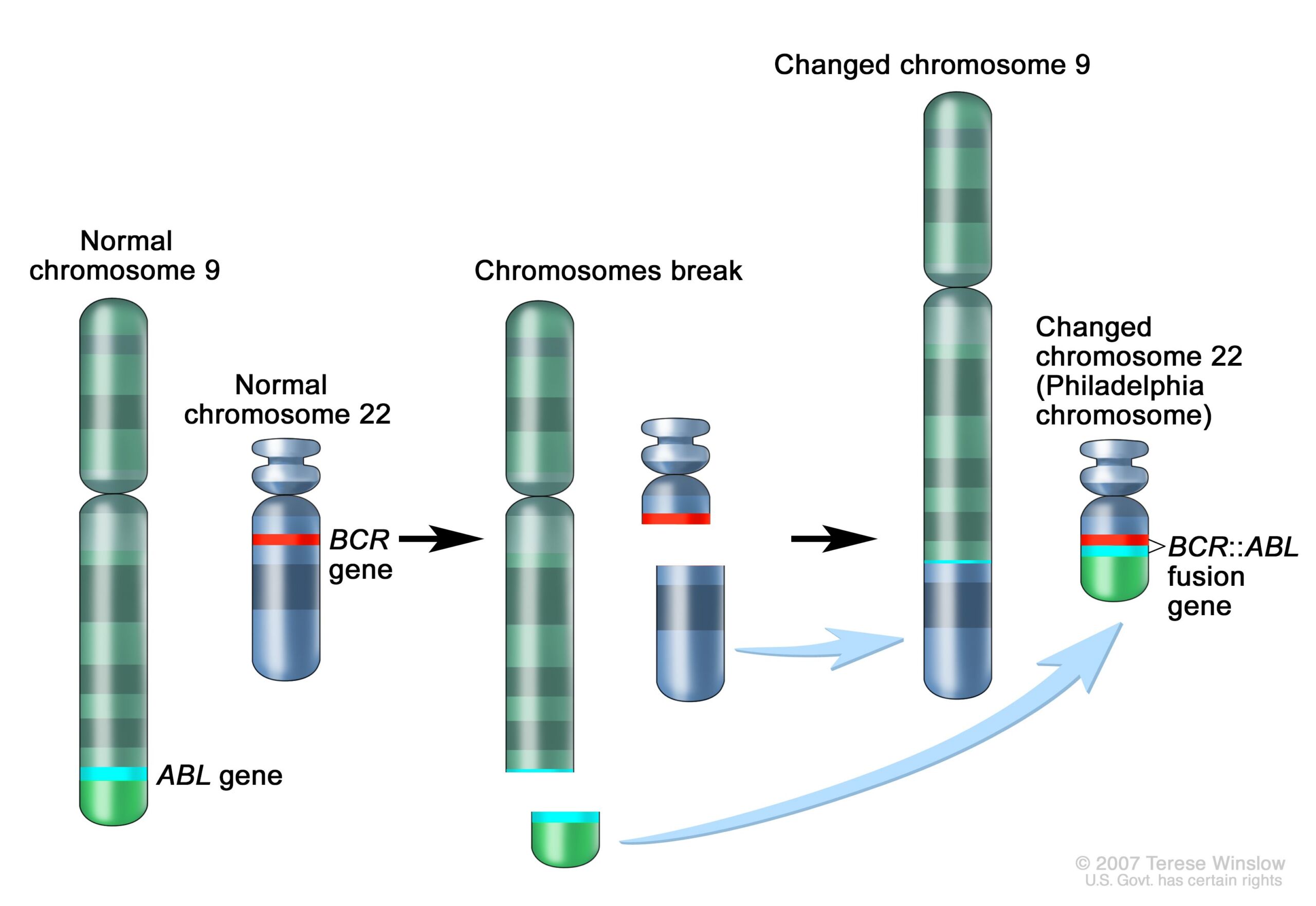
Later that same year, she saw another translocation, this time in patients with a different type of blood cancer, called chronic myelogenous leukemia (CML). Patients with CML were known to carry a puzzling abnormality in chromosome 22 that made it appear shorter than normal. The abnormality was called the Philadelphia chromosome after its discovery by two researchers in Philadelphia in 1959. But it wasn't until Rowley pored over her meticulously set dining table that it became clear why chromosome 22 was shorter—a chunk of it had broken off and traded places with a small section of chromosome 9, a 9;22 translocation.
Rowley had the first evidence that genetic abnormalities were the cause of cancer. She published her findings in 1973, with the CML translocation published in a single-author study in Nature. In the years that followed, she strongly advocated for the idea that the abnormalities were significant for cancer. But she was initially met with skepticism. At the time, many researchers considered chromosomal abnormalities to be a result of cancer, not the other way around. Rowley's findings were rejected from the prestigious New England Journal of Medicine. "I got sort of amused tolerance at the beginning," she said before her death in 2013.
The birth of targeted treatments
But the evidence mounted quickly. In 1977, Rowley and two of her colleagues at the University of Chicago identified another chromosomal translocation—15;17—that causes a rare blood cancer called acute promyelocytic leukemia. By 1990, over 70 translocations had been identified in cancers.
The significance mounted quickly as well. Following Rowley's discovery of the 9;22 translocation in CML, researchers figured out that the genetic swap creates a fusion of two genes. Part of the ABL gene normally found on chromosome 9 becomes attached to the BCR gene on chromosome 22, creating the cancer-driving BCR::ABL fusion gene on chromosome 22. This genetic merger codes for a signaling protein—a tyrosine kinase—that is permanently stuck in "active" mode. As such, it perpetually triggers signaling pathways that lead white blood cells to grow uncontrollably.

{kind=link}
By the mid-1990s, researchers had developed a drug that blocks the BCR-ABL protein, a tyrosine kinase inhibitor (TKI) called imatinib. For patients in the chronic phase of CML—about 90 percent of CML patients—imatinib raised the 10-year survival rate from less than 50 percent to a little over 80 percent. Imatinib (sold as Gleevec or Glivec) earned approval from the Food and Drug Administration in 2001, marking the first approval for a cancer therapy targeting a known genetic alteration.
With imatinib's success, targeted cancer therapies—aka precision medicine—took off. By the early 2000s, there was widespread interest among researchers to precisely identify the genetic underpinnings of cancer. At the same time, the revolutionary development of next-generation genetic sequencing acted like jet fuel for the soaring field. The technology eased the identification of mutations and genetic abnormalities driving cancers. Sequencing is now considered standard care in the diagnosis, treatment, and management of many cancers.
The development of gene-targeting cancer therapies skyrocketed. Classes of TKIs, like imatinib, expanded particularly fast. There are now over 50 FDA-approved TKIs targeting a wide variety of cancers. For instance, the TKIs lapatinib, neratinib, tucatinib, and pyrotinib target human epidermal growth factor receptor 2 (HER2), which runs amok in some breast and gastric cancers. The TKI ruxolitinib targets Janus kinase 2, which is often mutated in the rare blood cancer myelofibrosis and the slow-growing blood cancer polycythemia vera. CML patients, meanwhile, now have five TKI therapies to choose from.